Influence of microorganisms
MENU
Home
Categories
Classification
Bacteria
About Microscopic Organism
Microorganism
Definition
Function
Microbes
Types
Virus
Contact
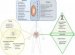
Bacteria Taxonomic Classification
Classification
/
Source:
RELATED POSTS
- E coli Taxonomy Classification
- Bacterial cells Function
- Bacteria and Viruses Similarity
LATEST POSTS
Functions of Pili in Bacteria
June 12, 2018
Protein Capsule Bacteria
December 5, 2018
Prokaryotes, Eukaryotes, and Viruses
February 15, 2018
Bacterial Infections
October 21, 2018
ADS BY GOOGLE
All you need is here
CATEGORIES
Classification
Bacteria
About Microscopic Organism
Microorganism
Definition
Function
Microbes
Types
Virus